

Understanding Cystic Fibrosis
Unlike other chronic lung diseases such as COPD, cystic fibrosis isn’t a disease that develops over time. Instead it is a lifelong, hereditary disease that affects your lungs, pancreas and other vital organs due to thick and sticky secretions.
The secretions are thin and slippery like a lubricant in a healthy patient, but a defective gene causes mucus and digestive juices to become thick and sticky.
Resulting in clogged airways and digestive passageways. When cystic fibrosis (CF) was first being diagnosed during the 1950’s, patients rarely reached the age to go to elementary school.

Progress is shown in treatments as in 1985 the average lifespan of patients was 25. As short ago as 2007 the diagnosis of cystic fibrosis meant patients still had a limited life span, but with cutting edge research efforts doctors have been able to extend the life expectancy of patients dramatically to a median age of 40.7 (cff.org).
With proper treatment and disease education, patients can still attend school, work and have a high quality personal life.
The difficulties cystic fibrosis poses for your lungs is mucus, which in a healthy person acts as a slimy lubricant, but the thick mucus associated with CF blocks the airways making breathing increasingly difficult which then results in serious and harmful lung infections. In 90% of patients with CF, the airways are affected. The pancreas is also harshly affected, the thick sticky mucus blocks the passageways to the digestive system which interferes with proper digestion and results in malnutrition.
Cystic Fibrosis Statistics
The following are statistics from the Cystic Fibrosis Foundation for the most recent patient data from 2013:
- 66% of New Cystic Fibrosis Diagnoses were in the First Year of Life
- 46% of Patients still Work Full or Part-Time
- 22% of CF Patients were Students
- In 2003 the Median Age of Survival was 33.4, in 2013 the Average is 40.7
- Nearly 50% of the CF Community is 18+

Who’s affected?
Cystic fibrosis isn’t more prevalent in male or females, both are at an equal risk of inheriting this genetic defect. While Caucasians experience the highest occurrence of CF at 86.8% of the total community, with only 4.3% of the community being African American and 7.9% being Hispanic in 2013. - Cff.org
Authoritative Resources
You may have a quick question that needs answered, but calling your doctor’s office and waiting for them to call back may take some time. To answer those questions or concerns quickly, you can visit one or all of the following authoritative CF resources:
- Cystic Fibrosis Foundation
- Cysticfibrosis.com (Great Blog Community Written by CF Patients)
- Daily Strength’s Cystic Fibrosis Support Group
- Cysticlife.org
The Causing Factor of Cystic Fibrosis
As previously said, cystic fibrosis is not a disease that develops after long-term exposure to smoking or other lung irritants. Instead it is a hereditary disease passed on from your parents. In the occurrence of CF, patients have a genetic mutation (defect) in a gene that causes a change in a protein that is responsible for adequately regulating the movement of salt in and out of cells. This then causes the normally slimy and thin mucus to become thick and sticky, which will then clog the respiratory, digestive and reproductive systems. While also increasing the concentration of salt in sweat. Many varying types of mutations can occur within the gene, but the type of gene defect is directly attributed to the severity of the disease.
Patients inherit the gene defect from both parents, which is a recessive gene, so the only way to develop cystic fibrosis is to inherit one copy of the gene defect from each parent. Only inheriting one copy of the mutation won’t lead to a development of CF, but you are still a carrier and have a chance of passing it on to your offspring.
Associated Symptoms
The felt symptoms of cystic fibrosis depend highly on the severity of the disease, you may even notice your regular symptoms worsen or get better with time. Which is why only some children experience related symptoms in their infancy, while others don’t begin experiencing symptoms until their teens or even adulthood.
Through advocacy and proven research, every newborn in all 50 states is now tested for cystic fibrosis before symptoms arise to provide treatment from day 1. Though if you were born before the regular newborn tests, it is important to recognize the symptoms and get tested sooner rather than later. One of the major signs of CF is an excessive level of salt in you or a loved one’s sweat. You’ll often be able to taste it when you give them a kiss. The rest of the symptoms are related to the respiratory and digestive systems.
Respiratory Symptoms
Due to the thick and sticky nature of your mucus attributed to cystic fibrosis, the tubes or airways that carry oxygen to and from your lungs become clogged. Resulting in the following symptoms:
- Persistent Cough that Results in Thick Mucus and Spit
- Breathlessness (Shortness of Breath)
- Wheezing
- Decreased Physical Ability
- A Regular Occurrence of Lung Infections
- Stuffy Nose
- Inflamed Nasal Passages
Digestive Symptoms
Not only does cystic fibrosis affect your respiratory systems ability to function properly, but the thick mucus of CF also affects and clogs the passageways that carry digestive enzymes from your pancreas to your small intestine. The lack of these enzymes inhibits your intestines ability to fully absorb important nutrients from the food you eat, leading to the following symptoms:
- Extreme Constipation
- Poor Growth and Slow Weight Gain
- Difficulty in Bowel Movements
- Commonly Experience Greasy or Bulky Stools
- Intestinal Blockage, Mainly in Newborns
The end of the large intestine, rectum, can protrude from the anus due to strain while passing stool (rectal prolapse). The occurrence of this in children may be a sign of cystic fibrosis, and parents should consult with a doctor about getting their child tested. Rectal prolapse in children may require surgery.
What's a CF Friendly Diet?
Living with CF is tasking, and as a CF patient you need a 25-50% higher caloric intake than a healthy person. You will also need 50% more protein than others your age. Additional calories shouldn't be consumed through sugary drinks or fatty foods. Instead you should cook fresh veggies, lean meats, chick, poultry and fish. Whether it's you or a loved one diagnosed, 3 meals a day with 3 snack in-between are generally recommend for a healthy disease conscious diet.
Known Risk Factors
- Family History – Being an inherited disease, cystic fibrosis is developed when the child inherits one recessive CF gene from each parent. Otherwise inheriting one set of the faulty gene will make them a carrier and potentially pass it on to their children. CF will not develop over time, it’s either you are born with it, born as a carrier or lack the mutated gene altogether.
- Race – Cystic fibrosis doesn’t strictly affect one specific race, but it is predominately seen in patients that are Caucasian with ancestors from Northern Europe.
Complications that May Arise with Cystic Fibrosis
The most common complications associated with cystic fibrosis occur in the digestive, respiratory and reproductive systems due to a build up of thick and sticky mucus. If you experience any of the following complications, you should immediately bring them up to your doctor to begin seeking treatment options before they escalate.
Complications Affecting the Respiratory System
- Bronchiectasis – This condition causes damage to your airways, which makes it increasingly difficult to fill and empty your lungs with oxygen due to the airways widening and becoming scarred and flabby. This occurs when the already damaged airways cannot be cleared of the thick and sticky mucus of cystic fibrosis.
- Symptoms: Coughing up high amounts of phlegm (sputum), tiredness and poor concentration, wheeziness, shortness of breath especially while exercising, recurring chest infections and coughing up blood.
- Treatment: Exercise therapy, physiotherapy, inhalers, immunization and hospital admission if symptoms are severe enough.
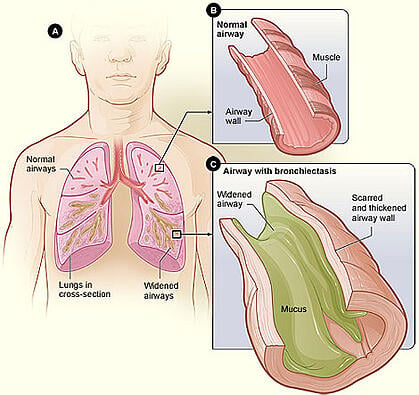
- Chronic Infections – Make sure to get a yearly flu and pneumonia vaccination to protect yourself from potentially damaging infections, as the thick built up mucus is a pristine breeding ground for harmful bacteria and fungi. Diagnosed patients experience bronchitis, pneumonia or sinusitis frequently throughout their life with CF, which can further increase the difficulty to breathe.
- Symptoms: Increased Wheezing or Shortness of Breath
- Treatment: Medication and antibiotics
- Preventative Measures: Get a yearly pneumonia and influenza vaccination, avoid crowded areas during peak flu season and wash your hands frequently.
- Coughing up Blood (Hemoptysis) – You may notice when coughing, mucus that is expelled is stained with blood. This is caused from the airways thinning from the effects of cystic fibrosis which is known ashemoptysis. The more severe the pulmonary impairment the more prevalenthemoptysis is, as you age you may notice it occurs more frequently that results in a further decline of lung function.
- Symptoms: Coughing up phlegm (sputum) that is tinted with blood
- Treatment: Antibiotics, or for severe cases of hemoptysis you doctor may recommend a bronchial artery embolization, surgical resection or bronchoscopic laser therapy.
- Nasal Polyps – This is when tissue forms and grows in the nasal passageways and sinuses due to the internal lining of the nose being inflamed and swollen. If you or a loved one has CF then the risk of this complication increases. Nasal polyps can be treated with nasal steroids, which will help reduce the often large polyps that can result in a runny or completely blocked nose. The large tissue growths can even result in a loss of taste and smell completely.
- Symptoms: Large growths that form in your nose of nasal cavity, nasal obstruction, nasal congestion, sneezing, runny nose, post-nasal drip, facial pain, loss of smell (anosmia), minimal ability to smell (hyposmia), loss of taste, itching around your eyes and chronic infections.
- Treatment: Nasal corticosteroid spray, oral corticosteroids, antibiotics or surgery.
- Collapsed Lung – With each lung infection your lungs become weaker and weaker, every infection takes more of a toll than the last leaving the lungs more likely to collapse (pneumothorax). Potentially resulting in requiring a transplant.
- Prevention: Get yearly vaccinations to reduce the occurrence of damaging infections.
- Treatment: Lung replacement surgery, oxygen concentrator.
- Respiratory Failure – Cystic fibrosis can damage your lung tissue to an extent that they no longer function. Your lung function will progressively get worse over time to potentially reaching a level that is life threatening. Oxygen therapy with a portable or home oxygen concentrator is an extremely beneficial treatment that you may want to discuss with your doctor.
- Symptoms: Shortness of breath, feeling like you can’t inhale enough oxygen, blueish colored skin, fingernails and lips.
- Treatment: Oxygen therapy or ventilator support.
- Pneumothorax – If you are experiencing heightened breathlessness and chest pain, pneumothorax may be the reason. Affecting older CF patients, pneumothorax is when air is collected in the area that separates the chest wall apart from the lungs. The collected air then begins to push against the outside of your lungs causing it to collapse, typically only a portion of the lung collapses.
- Symptoms: Shortness of breath, fast breathing, shallow breathing, chest tightness, chest pain and blue skin from a lack of circulation.
- Treatment: Pneumothorax is typically treated with either a chest tube or oxygen therapy.
Complications Affecting the Digestive System
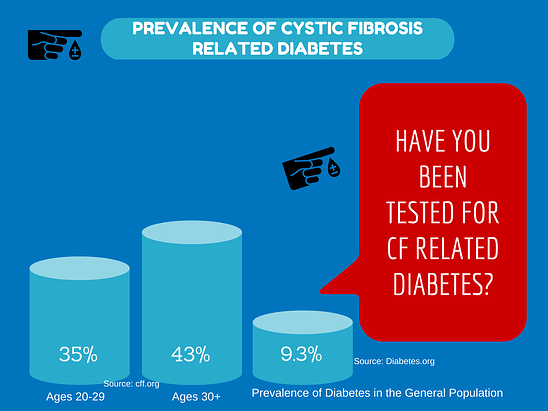
- Diabetes – According to the Cystic Fibrosis Foundation, 35% of patients aged 20-29 have cystic fibrosis related diabetes and 43% of patients 30 and older have diabetes related to CF (cff.org). Diabetes caused by CF is due to the pancreas, which produces insulin that allows your body to properly use and absorb sugar, becoming scarred due to the thick and sticky mucus that results in an inadequate amount of insulin production. As you age the chances of developing cystic fibrosis related diabetes increases.
- Symptoms: Increased thirst and urination, extreme fatigue, weight loss and an unexplained decline of lung function.
- Treatment: Doctors will use insulin to treat your CF related diabetes.
- Blocked Bile Duct – The bile ducts, which are responsible for delivering bile production from your liver helps your body digest fats and is stored in the gallbladder. Which becomes clogged and inflamed due to thick mucus blocking the bile from entering your small intestine. Resulting in liver problems, while gallstones have also been experienced.
- Symptoms: abdominal pain in the upper right region, dark urine, fever, yellowish skin, nausea and vomiting, or pale-colored stools.
- Treatment: Surgery to remove the gallstones from the bile ducts, or surgery to remove the gallbladder in all.
- Nutritional Deficiencies – Due to the thick and sticky mucus that is produced from cystic fibrosis, the digestive enzymes that allow your body to absorb vital nutrients from the food you eat that are produced in the pancreas are unable to reach your intestines. The thick mucus clogs the tubes that carry these important digestive enzymes to your intestines, which prevents your body from receiving proteins, fats or fat-soluble vitamins that are important for proper daily health.
- Symptoms: Lack of energy, feeling tired all the time, troubles gaining or losing weight, abdominal pain, gas and bloating.
- Treatment: Vitamin and mineral supplements.
- Intussusception – This is caused when part of the intestine is being pulled inward into itself, your intestine essentially folds into itself like an accordion. Intussusception is a high risk for children with CF, which can block food from passing through the intestine. If blood flow is blocked from parts of the intestine, those parts can die or heavy bleeding may also occur. If you or a loved one experiences intussusception, medical treatment should be immediately sought.
- Symptoms: Fever, bowl movements that are bloody and mucus-like, vomiting, blood and mucus mixed stools.
- Treatment: If the obstructed intestine cannot be reversed by an enema, surgery will be necessary.
- Rectal Prolapse – Due to frequent straining or coughing during times of constipation, the internal rectal tissue may protrude outside of the anus. In the event of a rectal prolapse you should call your doctor, majority of the time this can be fixed at home. This is most often seen in children and elderly patients.
- Symptoms: The main symptom is a reddish-colored mass sticking out of the anus.
- Treatment: Enemas, stool softeners, bulking agents, suppositories or surgery.
Additional Complications
- Osteoporosis – As the vital enzymes that absorb vitamins and minerals from food you digest are unable to reach the intestine through thick mucus, your bone strength suffers. Over time the depletion of bone building and strengthening nutrients such as calcium and vitamin D, will lead to the development of osteoporosis in patients with CF. This disease causes your bones to become fragile and weak, leaving your more susceptible to broken bones.
- Symptoms: Sometimes patients don’t experience any symptoms at all, felt symptoms can include back pain, bone fractures or loss of height.
- Treatment: Maximizing nutritional intake, daily exercise routine or for more sever causes your doctor may prescribe biosphosphonates to help influence new bone growth.
- Infertility – Thick and sticky mucus blocks the tube (vas deferens) that connects the prostate gland to the testes, which is why almost all males with a diagnosis of CF are infertile. Females are still able to reproduce, but pregnancy can heighten felt symptoms so you should first speak with your doctor about the risks of getting pregnant.
- Electrolyte Imbalances – As your body isn’t able to properly regulate the amount of minerals in your body, there is an excess level of salt in your sweat which can lead to an imbalance of minerals in your blood.
- Symptoms: Increased heart rate, fatigue, low blood pressure, vomiting, diarrhea and weakness.
- Treatment: Providing specific electrolyte replacements, intravenous fluids, diet adjustments and mineral supplements.
Treating Your Cystic Fibrosis
Currently there is no definitive cure for cystic fibrosis, though there are ample amounts of treatment avenues your doctor can take that will reduce felt symptoms so you can be a more active, healthier and happier you. Treatments can vary from medications to specific therapies that target and strengthen your respiratory system. Depending on your exact condition your doctor may recommend one or many of the following CF treatment options.
Treatment Goals:
- Prevent the Occurrence and Reoccurrence of Lung Infections
- Expelling and Breaking Apart Thick and Sticky Mucus in the Lungs
- Provide Patients with Adequate Nutrition Information
- Treating Intestinal Blockage and Preventing it from Returning
Standard Medications
Typical medications your doctor may prescribe to treat CF include:
- Antibiotics – These are used if you have developed a threatening lung infection, while they are also used in the prevention of lung infections.
- Drugs that Thin Mucus – Which break apart the thick layers of mucus clogging up your lungs, which will make coughing the mucus up easier and resulting in an improved lung function.
- Bronchodilators – This treatment method relaxes the tightened muscles around the bronchial tubes to open up the airways.
- Oral pancreatic Enzymes – The vital enzymes produced in the pancreas are necessary to absorb vital nutrients and minerals from the food you eat are blocked from reaching the digestive tract. Oral pancreatic enzymes will help your digestive tract absorb these extremely vital nutrients to help protect you from nutritional deficiencies.
Chest Therapy
To help loosen up the thick and sticky mucus in your chest, your doctor may recommend a few chest physical therapies that will make it easier for you to cough up and expel mucus from your body. A popular and highly used technique you doctor will talk about is chest clapping, which is where you “clap” both the front and backside of your chest with cupped hands to help loosen up mucus. For the best results, this should be done 1-4 times per day.
In addition to clapping with cupped hands on your back to break apart mucus, mechanical devices have also been developed and used to help loosen mucus in your chest which include:
- The Chest Clapper – Just like when using cupped hands to clap on the front and backside of your chest to free sticky mucus, the chest clapper is a handheld mechanical device that replicates cupped hands clapping on the chest.
- Inflatable Vest – An inflatable vest that fits around your chest and vibrates at a high rate. Which will shake and loosen the thick and clogged mucus in your lungs to simplify breathing.
- Breathing Devices – Your doctor will have you use a tube or mask to breathe through while doing different breathing exercises. Which will loosen the thick, sticky lung mucus so it can be cleared easier with coughing. Clearing the mucus from your lungs will reduce the occurrence of harmful lung infections, while also improving lung function.
Pulmonary Rehabilitation Treatments
One of the most beneficial tools for patients with cystic fibrosis that your doctor may or probably has recommended is getting enrolled in a pulmonary rehab program. These programs are typically on an outpatient basis and are long-term, during the program you will be equipped with tools and knowledge that will improve your stamina, overall happiness and most importantly lung function. During the entire program you will have a dedicated team of medical specialist to answer any questions or concerns you may have along the way. Pulmonary rehab programs include:
- Exercise Training
- Techniques to Conserve Energy
- Nutritional Counseling
- Breathing Techniques
- Group Support, Psychological Counseling or a Combination of Both
Benefits of Pulmonary Rehabilitation:
- Greatly Improves Your Overall Quality of Life
- Function Better in Day to Day Life
- Heightened Ability to Exercise
- Decreases the Felt Symptoms of Cystic Fibrosis
- Gives You the Tools to Better Manage Stress and Anxiety

Additional Treatment Options
- Nasal Polyp Removal Surgery – If nasal steroids, oral corticosteroids or other treatment options are unsuccessful in removing your nasal polyp your doctor may recommend surgery to effectively remove the polyp.
- Oxygen Therapy – As cystic fibrosis affects the lungs, it can hamper your lungs ability to produce and deliver oxygen throughout your body to the vital organs that results in shortness of breath (breathlessness). If your blood oxygen levels are at a low enough level your doctor will prescribe supplemental oxygen through oxygen therapy. You may be thinking of bulky and cumbersome oxygen tanks, but with the advancements in oxygen therapy those bulky tanks are replaced with lightweight portable oxygen concentrators. Which allow you to run errands, travel and anything else you set your mind to while staying properly saturated. Portable oxygen devices don’t need to be refilled or replaced, instead they are powered by rechargeable lithium ion batteries. Your doctor may recommend all day use of an oxygen concentrator or just for use during times of peak physical activity. Oxygen therapy is also used to reduce the occurrence of high blood pressure in your lungs that is known as pulmonary hypertension.
- Endoscopy and Lavage – Using an endoscope, your doctor will suction out mucus from your obstructed airways to improve your breathing function.
- Feeding Tube – Malnutrition is a common occurrence with cystic fibrosis as your body is unable to absorb the full amount of vital nutrients your body needs to function properly due to mucus blockage of the intestines. Your doctor may recommend using a feeding tube at night while you sleep to supplement the lack of nutrients. A feeding tube is either inserted through your nose to your stomach or a tube is surgically implanted into your abdomen.
- Lung Transplant – If your respiratory problems keep increasing, you experience severe breathlessness or your body is developing a resistance for antibiotics that used to fight off infections. Or if other treatment options have begun to fail or your shortness of breath gets extreme, a lung transplant may be recommended by your doctor. When receiving a lung transplant, you will have to replace both because cystic fibrosis doesn’t affect only a single lung.
- Bowl Surgery – If you are experiencing a blockage in your bowels and other treatments have failed, your doctor may talk to you about undergoing bowl surgery to remove the blockage.
WOW! What an incredible amount of useful information. Now it’s up to you what you do with it. Living with cystic fibrosis will be tasking to say the least, but by following the information provided you can live an active, healthy and most importantly a happy lifestyle. By following our preventive measures and staying away from symptomatic triggers, cystic fibrosis can be controlled to an extent that it won’t consume your entire life. Do you have some tips on how to live the highest quality of life possible with CF? If so leave a comment below, you never know the impact your tip may have on someone else struggling with CF.

